




摩登7平台合作客户/
拜耳公司 |
同济大学 |
联合大学 |
美国保洁 |
美国强生 |
瑞士罗氏 |






相关新闻Info
推荐新闻Info
-
> 不同PQAI溶液静态/动态表面张力变化及对脉动热管性能影响(三)
> 不同PQAI溶液静态/动态表面张力变化及对脉动热管性能影响(二)
> 不同PQAI溶液静态/动态表面张力变化及对脉动热管性能影响(一)
> 界面流变仪可以测量液体表面张力吗?界面流变仪与界面张力仪区别解析
> 测量表面张力/界面张力的仪器有哪些?怎么选
> PG脱酰胺添加量对玉米醇溶蛋白气-水动态表面张力的影响
> 摩登7表面张力仪使用指南【专业版】
> 平面流动皂膜表面张力系数、厚度和流动速度实验装置及测量方法(二)
> 平面流动皂膜表面张力系数、厚度和流动速度实验装置及测量方法(一)
> 单层膜界面上亚微米颗粒表面张力阻力系数修正——颗粒在单层膜上的阻力系数
探索泡沫粗化与表面流变学之间的关联性疏水性蛋白——结果和讨论
来源:上海谓载 浏览 2568 次 发布时间:2021-11-25
结果和讨论
泡沫稳定性与气泡尺寸演变
我们已按照上一节所述的程序制备了泡沫,并对所有材料进行了研究。 值得一提的是,HFBII、Quillaja皂甙和b-酪蛋白的发泡溶液含有0.1 wt%的发泡剂,而b-lg的发泡溶液含有0.5 wt%的材料,以达到所需的空气体积分数。 通过这种方式,我们确保气泡完全由所选发泡剂以最佳方式稳定。 随后将这些泡沫转移到0.5 wt%的黄原胶溶液中,提供足够高的屈服应力,以防止直径小于200 mm的气泡在大约2周内形成泡沫乳状物(即液体排出)。31最终泡沫样品含有大约50 vol%的空气,低于球体的紧密堆积密度, 确保气泡之间最小的薄膜形成和聚结。 所有这些使我们能够排除聚结和排水的影响,并将注意力集中在泡沫中气泡的歧化上。 使用浊度扫描测量设备,我们观察到所研究系统中随时间变化的平均气泡尺寸演变,d(t)/d(0),如图1所示。 从数据可以清楚地看出,在实验的时间范围内(即2周),由HFBII稳定的泡沫没有显示出明显的粗化。 由b-酪蛋白稳定的泡沫粗化速度快得多,约为40分钟,而b-lg和Quillaja皂甙粗化时间约为70分钟。 在显著粗化后,泡沫中的气泡变得过大,其浮力无法被生物聚合物基质的屈服应力抑制,导致在几天内形成奶油状、随后的聚结和泡沫崩塌。
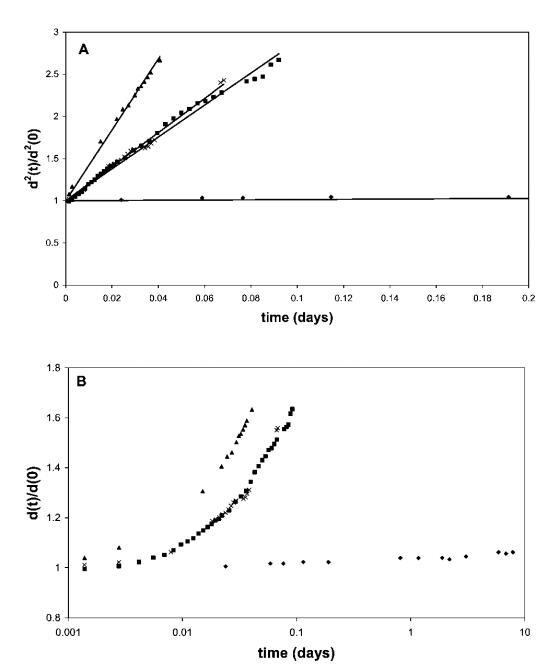
图1面板A显示了黄原胶溶液中50 vol%气泡的相对平方气泡直径d2(t)/d2(0)。 泡沫由含有0.1 wt%HFBII(A)、0.5 wt%b-lg(-)、0.1 wt%b-酪蛋白(:)和0.1 wt%Quillaja皂甙(*)的水溶液生成。 将所得泡沫转移到0.5 wt%黄原胶溶液中。 面板B以对数时间刻度显示整个时间间隔内的数据。
该结果与先前报道的稳定性试验结果一致,26表明HFBII稳定的泡沫粗化时间与牛奶蛋白质或皂甙的粗化时间之间存在几个数量级的差异。 由于泡沫的体积分数低于气泡的紧密堆积,并且最初抑制了奶油化的影响,因此泡沫稳定性的这些巨大差异归因于歧化率的差异。 在所有情况下,我们在相同条件下使用相同的气体成分(空气)(即气体溶解度相同),歧化率的差异与空气/水界面处吸附乳化剂层的表面流变特性有关。 在此背景下,值得一提的是,尽管b-lg和奎拉叶皂甙的泡沫粗化行为非常相似,但奎拉叶皂甙泡沫的整体稳定性(和总寿命)更好。 然而,这应归因于干燥泡沫中皂甙稳定的薄液膜的更高稳定性,该液膜在后期由于液体排出和大气泡奶油化而形成。
此外,从这些数据可以清楚地看出,除HFBII外,所有系统的视气泡半径都发生了显着变化,这对应于单个气泡的空气/水面处的极大表面变形。 考虑到蛋白质吸附可能是不可逆的,这意味着在歧化发生的大部分时间内,表层处于大变形阶段,并且与相的大部分不平衡。 这不同于通常提取表面模量并随后假设常数的小变形区域。 3,9根据上述观察和推理,我们继续研究空气/水界面扩展层的大变形行为。 考虑到蛋白质吸附可能是不可逆的,这意味着在歧化发生的大部分时间内,表层处于大变形阶段,并且与相的大部分不平衡。这不同于通常提取表面模量并随后假设常数的小变形区域。3,9根据上述观察和推理,我们继续研究空气/水界面扩展层的大变形行为。
大变形膨胀表面流变学
狂犬病HFBII。 图2a显示了HFBII的3个压缩等温线的典型序列。 曲线中的每个压缩可分为三个部分。 在低表面压力下,当蛋白质分子处于非常稀释的“气体”状态时,表面压力的增加可以忽略不计。 当压力开始增加,分子失去空间自由度(液相)时,弹性也逐渐增加。 如果层被完全压缩,则会观察到曲线中的弯曲点。 考虑到图中表示表面单层的第一个弯曲点,在约20 mN m-1的表面压力下,该点处每个HFBII分子的表面积约为400 a˚2。 假设球形六边形紧密堆积,每个分子的面积相当于蛋白质直径约2.25 nm,这与根据晶体结构确定的分子大小(约2.5 nm)相当好。3
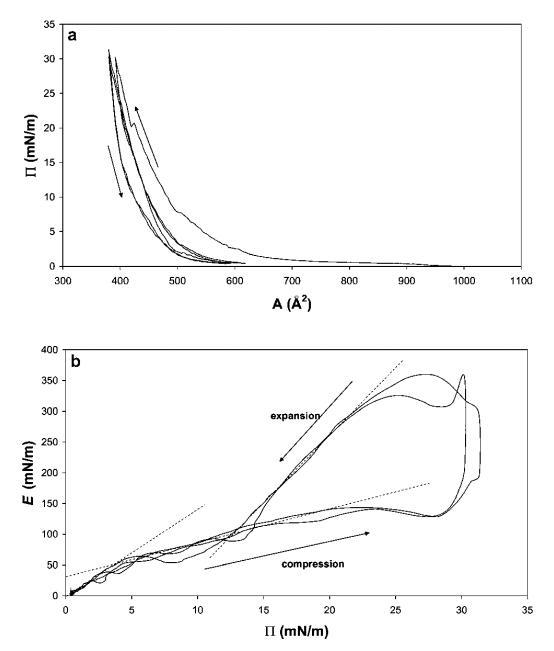
图2(a)15 mg HFBII在中性pH水上扩散的P–a和(b)E–P曲线,根据相同数据在20℃下测量。 下面板中绘制的线表示不同的状态。
Paananen等人37测量了HFBII的类似压力-面积曲线,他们研究了两种II类疏水蛋白HFBI和HFBII分子膜的结构层次。 报告的工作不同于我们的实验,因为他们使用pH值为5.0的醋酸盐缓冲液作为亚相。 与本文给出的结果相比,接近HFBII pI的pH条件可以解释每个分子的面积略小。
当表面层膨胀时,观察到明显的滞后效应,即压力下降比压缩时快得多,最后下降到几mN m-1,与压缩前类似。 在第二次压缩时,在与第一次压缩相似的压力下,每个分子的面积通常稍小,这表明在第一次压缩期间分子在表面发生了一些重排。 如果该层被完全压缩,则可在20 mN m-1的表面压力周围看到薄膜屈服。 该屈服点在图3后面的部分中更为明显。

图3 HFBII的代表性压缩等温线,压缩量远低于每个分子的面积。 (A) P(-)和E(---)与每个分子的面积的关系。 (B) 摊铺量和压缩(Gcomp,-)预期的表面载荷,并通过椭偏仪(Gell,A)测量。
在图2b中,根据表面压力P绘制了模量E。可以观察到三种不同的状态,与上面讨论的类似。 压缩后,在低P时,观察到约14的坡度,在Ⅱ=4 mN m-1处,有一个拐点朝向较低的坡度。 该拐点反映了E随P增加的机制变化。这可能是由HFBII分子形成的松散的自组装2D网络引起的,对于弱吸引力粒子也是如此。 第二种状态持续到20 mN m-1的表面压力。 在左面板的拐点之外,可以观察到模量再次开始上升,并具有非常高的斜率。 在这里,2D蛋白质网络可能已经被压缩成一个几乎紧密堆积的单层,其中单个HFBII分子的硬度再次被反映出来。
在屏障方向迅速恢复的点处,观察到E跳变,这很可能是由于界面适应变形方向变化的时间有限而产生的流体动力效应。 一般来说,经历短时(高频)变形的材料没有时间自然重建/修复其内部结构,这可能导致薄膜破裂。38
随后,在松弛过程中,模量重新连接到压缩曲线,表明弹性蒙皮的松弛和重组。 在低于15 mN m-1的压力下,该网络可能再次开始可逆地重新排列,以便分解为低于Ⅱ=5 mN m-1的单个实体。
图3显示了远高于单层过渡的代表性压缩等温线。 在每分子面积以下进一步压缩时,表观表面压力降至约60 mN m-1,这是非常高的。 该层的强度也反映在143 mN m-1的表观表面膨胀模量(Eapp)峰值中。 我们必须记住,在这些高表面压力下,由于表面形成凝胶蛋白质层,表面压力的测定可能变得不准确。 因此,除了表面流变参数外,图3B中还提供了椭偏仪数据,这些数据是通过使用实验部分所述的Multiskop椭偏仪装置在朗缪尔槽的空气/水界面进行同步椭偏仪测量获得的。
椭偏仪数据中,我们可以使用de Feijter的方法在压缩过程中的每个点获得蛋白质吸附凝胶(t)量的估计,因为该过程足够慢,允许我们进行椭偏仪测量。如果蛋白质在整个压缩过程中仍不可逆地吸附在界面上,则凝胶(t)应根据Gcomp(t)=M0/A(t))遵循表面积的变化,其中M0是界面处蛋白质的初始量,A(t)是槽屏障之间的面积。
如图3B所示,两条曲线实际上与表面压力值一致,约为Ⅱ=5 mN m-1。在这一点之后,Gell突然增加,其值比假设所有蛋白质在界面上均匀分布的估计值高出两倍。这种行为表明蛋白质层是不均匀的,或者空气/水界面不是平坦的,这是由于界面上形成了微观褶皱。事实上,在压力超过30 mN m-1时,肉眼可以看到界面皱纹的外观,并且可以在不放大的情况下看到。这些结构的出现开始于移动屏障附近,并随着压缩的进行而传播。为了获得更好的图像,我们在表面压缩和膨胀期间直接进行了光学显微镜观察(图4),使用底部带有蓝宝石窗口的朗缪尔槽,将其置于透射光学显微镜的顶部。如图5所示的垂直线(在不同放大率下)平行于屏障对齐。
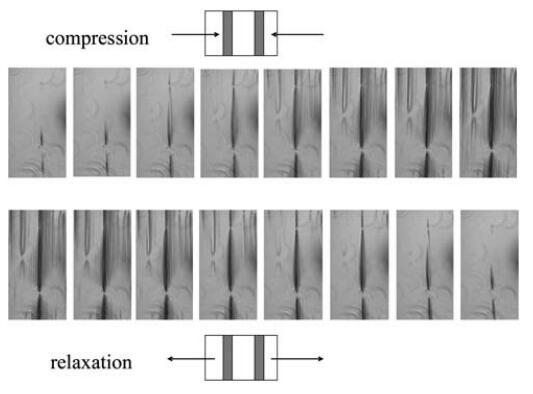
图4在4倍放大的透射光显微镜下,扩展HFBII层的压缩和松弛。

图5在透射光显微镜下,疏水蛋白在40 mN m-1下的压缩。左侧放大4倍,右侧放大10倍。
压缩时结构的形成和表面积松弛时结构的消失是可逆的,类似于表面性质P、E和凝胶。当在较高放大率下观察显微图像时,观察到皱纹的均匀空间分布约为4mm。当将椭偏仪数据与显微图像进行比较时,我们观察到椭偏仪信号在比观察到皱纹的地方低得多的表面压力下增加。这可能意味着起皱发生在比显微镜下观察到的小得多的长度尺度上。然而,皱纹的初始分布是相当不均匀的,没有皱纹的区域和皱纹出现的区域具有恒定的空间分布。这种行为与最近发表的Kralchevsky等人的弹性朗缪尔层理论39的预测一致,也可能解释椭偏测量中的噪声。
藜芦皂甙。奎拉叶皂苷的压缩曲线如图6所示。该皂甙在拐点处显示出非常高的表面压力,最大表面压力为45 mN m-1。此外,皂甙表现出很宽的迟滞回线。从E–P曲线推断出斜率为4.3,观察到E中出现2个平滑的峰,这可能表明界面中存在相变,从皂甙的相对拉长的疏水区域判断。27当屏障方向逆转时,观察到E中存在较大的超调,表明膜破裂,类似于HFBII。
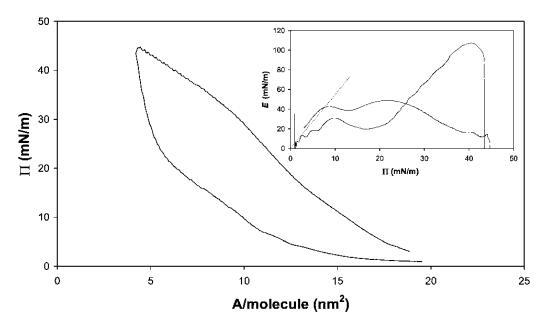
图6在20℃下测得的30 mg奎拉叶皂苷在中性pH水上的P–A和(插图)E–P曲线。插图中绘制的虚线表示E–P曲线的初始坡度。
b-乳球蛋白。图7显示了b-乳球蛋白的压缩等温线。这条曲线远没有HFBII曲线陡峭,拐点更为明显。尽管在屏障运动逆转时观察到滞后现象。E–P曲线的初始斜率为4.3,远小于低P区的HFBII,表明b-lg分子在界面中相对较软。观察到E的平滑最大值为43 mN m-1,之后在进一步压缩至28 mN m-1时,不再检测到E的恢复。结果与早期的研究结果一致。40,41在屏障逆转时,观察到E的超调,再次表明结构无法适应突然膨胀和断裂。

图7在20℃下测量的15 mg b-乳球蛋白在中性pH水上的P–A和(插图)E–P曲线。 插图中绘制的虚线表示E–P曲线的初始坡度。
图8显示了被吸附的b-乳球蛋白层的压缩曲线,在这里,同时进行了椭偏测量。 与HFBII类似,Gell最初遵循Gcomp。 压缩一段时间后,Gell开始变得比Gcomp小,这表明存在不均匀层或某些材料从表面解吸。 进一步压缩和膨胀后,凝胶仍小于Gcomp,这表明解吸或表面聚集。 这与其他地方对吸附b-lg层的类似发现一致。42

图8代表性压缩-吸附b-乳球蛋白(c=0.01 wt%)的膨胀实验,压缩低于每个分子的面积。 通过扩散量和压缩量(Gcomp)和椭偏仪(Gell)测量的预期吸附量。
酪蛋白。 对于纯b-酪蛋白,再次观察到不同的行为(见图9)。 等温线的形状更平滑,没有观察到滞后现象。 观察到的斜率E/P为6.5,与之前报告的吸附层结果一致。43 E–P曲线表明,E的最大值约为25 mN m-1,之后观察到下降。 该结果与Rodriguez-Patino等人44和Hotrum等人41的早期测量结果一致。此外,E中并未明显反映出势垒方向的反转,这表明结构的松弛速度与势垒反转速度一样快,即约1秒。
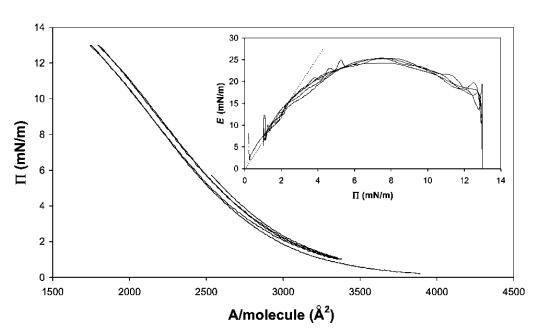
图9 8 mg b-酪蛋白在水中的P–A和(插图)E–P曲线,神经pH值,在20℃下测量。 插图中绘制的虚线表示E–P曲线的初始坡度。
频率依赖性。 为了评估其中一些系统中粘性损失的重要性,我们对b-lg和HFBII进行了频率相关的表面膨胀流变。 对于表面膨胀流变学,我们显示了在18<P<22 mN m-1之间测量的小变形模量(见图10)。 注意,变形率d(lna)/dt为0.0012 s 1对应于5 mm min 1的势垒速度,用于大变形实验。 相比之下,歧化时间约为0.5小时,接近泡沫实验观察到的粗化时间。 HFBII的数据表明,在所研究的变形速率范围内,变形速率对E的影响适中。 对于b-乳球蛋白,频率的影响更为明显,因此,在变形速率低于0.001 s 1时,可观察到E的强烈下降。 这表明b-lg的粘性损失比HFBII更为明显。 这一比较再次表明,b-lg的外观更柔软,压缩/膨胀时解吸/吸附能力更强。
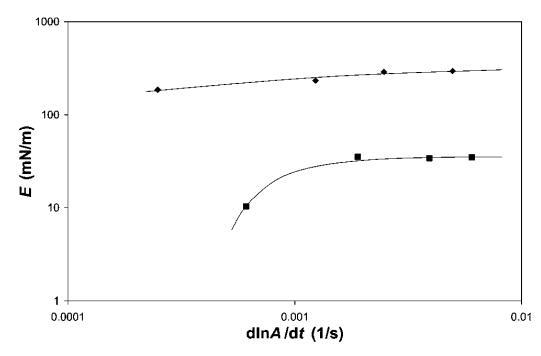
图10 HFBII(A)和b-lg(-)的表面膨胀模量E,通过18和22 mN m-1之间的线性压缩-膨胀环测量。 画线来引导眼睛。
表面剪切流变学。 所有四个系统的表面剪切流变测量如图11所示。 我们观察到HFBII具有模量,比b-乳球蛋白和皂甙高一个数量级,这与早期结果一致。25值得一提的是,对于像奎拉叶皂甙这样的表面活性剂,测得的表面剪切弹性非常高。 b-酪蛋白的表面剪切模量要低一个数量级。 关于频率,我们发现与HFBII相比,皂甙和b-乳球蛋白对频率的依赖性更强,尤其是在低频率下。 此外,对于HFBII而言,Gs 00/Gs0的比率是最低的,即表面弹性对模量的贡献比表面粘度大得多。 当我们向交叉点外推时,我们预计HFBII和b-乳球蛋白或皂甙之间的弛豫时间会有一个数量级的差异。 对于b-酪蛋白,从0.01 Hz左右的数据可以很容易地观察到交叉点。
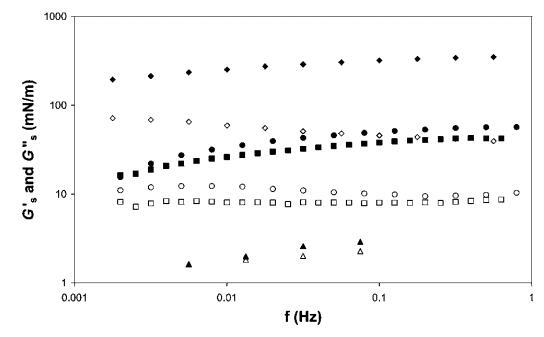
图11 HFBII(a)、b-乳球蛋白(-)、b-酪蛋白(:)和奎拉叶皂苷(C)的表面剪切储存(填充符号)和损失模量(开放符号)作为振荡频率的函数。
一般性讨论
表1列出了从E–P曲线、表面剪切流变模量和泡沫粗化时间获得的参数汇总。 我们还用幂律拟合了E/P等温线的初始部分。43这使得我们可以在分子间相互作用可以忽略的(半)稀释条件下对2D均聚物的行为进行类比,并且使用标度参数可以得出(界面处2D聚合物的)回转半径Rg 将线圈中的分段数缩放为Rg f Nn。 结合模量和表面载荷的定义,得出表面压力和表面膨胀模量E=yP之间的直接关系,其中y与n的关系为n=½y/(y 1)。 对于2D中的自回避线圈,即膨胀聚合物链,n等于0.75,而对于Q条件,n等于0.57。 对于折叠线圈,对于折叠分子或硬盘,n进一步降低至0.5。45我们将使用此框架将所考虑的实验系统的表面行为映射到具有类似膨胀模量与表面压力依赖关系的等效2D聚合物上。 这将使我们更容易进行比较,并得出一些非琐碎的结论。
表1在20℃和中性pH条件下进行的膨胀实验中研究的材料的典型参数。Pmax和Emax表示最大表面压力和表面膨胀模量。 幂律指数y等于E与P的斜率,n是2D线圈半径与其分段数的结果缩放指数

首先,在表观分子“硬度”方面,可以看出明显的差异。 HFBII具有更高的斜率y,因此v值明显较低。 这表明在稀释状态下,HFBII表现为等效的紧密折叠聚合物链,而牛奶蛋白在中等质量溶剂中表现为等效的2D聚合物链。 这与蛋白质结构的差异是一致的。 一方面,HFBII具有相对较小的分子质量和四个二硫键,形成一个刚性致密球体,据信在吸附时不会明显展开。23另一方面,b-lg的分子质量较大,二硫键较少。46此外, 经常有人认为,b-lg在吸附时展开。47,48这些特征结合在一起,使分子在空气-水表面扩散时具有更大的灵活性。
同样值得一提的是,在这个温度下,b-lg和b-酪蛋白之间的斜率有一个小但显着的差异。 虽然这两个值都表明了q状态和自回避行走之间的情况,但b-lg聚合物的等效物似乎比b-酪蛋白的自回避程度略高。 在环境温度下,Hambardzumyan等人认为,b-酪蛋白分子之间的范德华力或氢键等吸引性相互作用很重要。 43这可能解释了与b-lg相比,b-酪蛋白的结构更紧凑,表明b-酪蛋白分子间的相互作用更强, 38因此,由于蛋白质结构的这些差异,不同的蛋白质在压缩时在表面流变性方面可能表现出很大的差异。 虽然这两个值都表明了q状态和自回避行走之间的情况,但b-lg聚合物的等效物似乎比b-酪蛋白的自回避程度略高。在环境温度下,Hambardzumyan等人认为,b-酪蛋白分子之间的范德华力或氢键等吸引性相互作用很重要。43这可能解释了与b-lg相比,b-酪蛋白的结构更紧凑,表明b-酪蛋白分子间的相互作用更强,这也与其类似于二嵌段共聚物的分子结构相一致。38因此,由于蛋白质结构的这些差异,不同的蛋白质在压缩时在表面流变性方面可能表现出很大的差异。
最后,等效“聚合物”具有与稀释奎拉叶皂苷层相同的E/P等温线,在表面表现为相当软的实体,这可能是由于其相对较大的每分子面积与分子质量比。27
除了为稀释表面提供的参数外,还可以从较大压缩下的数据中获得更多定性信息。 对于HFBII而言,压缩后模量持续增加,这可视为一种膨胀硬化。 虽然在单层转变之后,表观模量降低,但我们必须意识到,E的下降可能不是真实的。 由于起皱,每个分子的面积不再发生真正的变化,尽管障碍物仍在移动。 此外,由于褶皱,靠近探针的弯月面可能变得不规则,导致P测量中出现伪影。 对于所有其他分子,可以观察到膨胀软化,表明软分子被压缩或部分分子从表面排出。 因此,各层在压缩后逐渐变弱。 因此,在歧化过程中的某一点之后,随着系统阻力的降低,气泡收缩率将显着增加。 这与在工艺早期观察到的情况完全相反,在该过程中,气泡收缩引起的表面积小幅度减少会导致表面膨胀模量增加,从而加强该层并增加其抗歧化能力。 此外,在大多数理论考虑中,未考虑此类行为,表面模量根据小变形实验获得的斜率或使用给定平衡吸附值的吉布斯弹性值作为常数。 对于HFBII而言,压缩后模量持续增加,这可视为一种膨胀硬化。虽然在单层转变之后,表观模量降低,但我们必须意识到,E的下降可能不是真实的。由于起皱,每个分子的面积不再发生真正的变化,尽管障碍物仍在移动。此外,由于褶皱,靠近探针的弯月面可能变得不规则,导致P测量中出现伪影。对于所有其他分子,可以观察到膨胀软化,表明软分子被压缩或部分分子从表面排出。因此,各层在压缩后逐渐变弱。因此,在歧化过程中的某一点之后,随着系统阻力的降低,气泡收缩率将显著增加。这与在工艺早期观察到的情况完全相反,在该过程中,气泡收缩引起的表面积小幅度减少会导致表面膨胀模量增加,从而加强该层并增加其抗歧化能力。此外,在大多数理论考虑中,未考虑此类行为,表面模量根据小变形实验获得的斜率或使用给定平衡吸附值的吉布斯弹性值作为常数。
从b-乳球蛋白的椭偏仪数据来看,后者确实如此(见图8)。 从膨胀和剪切作用下的频率行为可以看出,分子柔软度和吸附能都很重要。
我们现在想将表面流变参数转化为对泡沫中歧化稳定性的理解,并给出2Emax/gmax作为这一点的度量。 我们进一步注意到,根据Kloek等人的计算机模拟,在预测气泡收缩时,2Emax/gmax应大于5而不是1.2,我们假设表面变形为纯弹性。 然而,对于几乎所有的材料,变形都是弹性和粘性作用的复杂量。
从频率相关的表面膨胀和剪切流变学可以得出结论,HFBIIlayer的变形比Quillaja皂甙、b-乳球蛋白或b-酪蛋白层的变形更具弹性。 关于大变形、气泡收缩和歧化,这表明HFBII的非平衡模量比其他材料的非平衡模量更受弹性贡献的支配。
当我们现在再次考虑表1中的2Emax/gmax比率时,很明显HFBII超过了5的值,这表明这将稳定泡沫,防止收缩。 根据Kloek等人的说法,对于b-乳球蛋白和皂甙,我们报告的值介于1和2之间,表明气泡收缩延迟,但没有完全停止。2食品蛋白质通常属于这种状态,对歧化过程的抵抗力相对较小。10最后, 根据这个比率,b-酪蛋白根本无法抵消气泡收缩,因为2Emax/gmax<1,这是主要的吉布斯标准。
现在,我们将把表面流变学数据与泡沫的粗化时间以及早期报告9、25、26进行比较,其中探讨了单个泡沫和泡沫的寿命。 我们发现HFBII稳定的泡沫或气泡比其他所有泡沫或气泡的寿命更长。 考虑到所研究表面层的全压缩等温线、2Emax/gmax比、松弛行为和表面剪切数据,我们发现表面膨胀实验对气泡抗收缩稳定性具有良好的预测能力。 更准确地说,可以看出,与其他系统相比,HFBII在所有膨胀量方面都非常突出,因此提供了一个独特的机会,可以在更广泛的泡沫寿命范围内进行比较。 我们发现b-酪蛋白的最小粗化时间与它不符合吉布斯标准这一事实密切相关。 最后,b-lg和Quillaja皂甙的粗化时间相似,但它们比b-酪蛋白的粗化时间大。 同样的趋势也适用于表面膨胀参数,它们确实超过了吉布斯准则,但并不远。 因此,这些物质可以减慢,但不能阻止歧化。
与表面膨胀流变学一样,表面剪切模量似乎也给出了与tcoarse相同的趋势。 然而,从理论上看,膨胀而非剪切是与气泡收缩相关的主要变形。 因此,可以预期歧化率与膨胀模量之间存在直接联系,而与剪切力之间存在间接联系,因为通常所有表面参数都是表面吸附的相互依赖和单调函数。
最后,我们假设,如果一个吸附的蛋白质层既满足2Emax/gmax>5的标准,又表现出有限的粘性损失,那么它可以在数月的时间尺度上有效地阻止歧化。
探索泡沫粗化与表面流变学之间的关联性疏水性蛋白——摘要、介绍
探索泡沫粗化与表面流变学之间的关联性疏水性蛋白——材料和方法
探索泡沫粗化与表面流变学之间的关联性疏水性蛋白——结果和讨论
探索泡沫粗化与表面流变学之间的关联性疏水性蛋白——结论、致谢!